Спинальная мышечная атрофия
| Спинальная мышечная атрофия | |
|---|---|
 | |
| МКБ-10 | G12. |
| МКБ-10-КМ | G12.1 и G12.9 |
| МКБ-9 | 335.0-335.1 |
| МКБ-9-КМ | 335.1 и 335.10 |
| OMIM | 253300 |
| DiseasesDB | 14093 |
| MedlinePlus | 000996 |
| MeSH | D009134 |
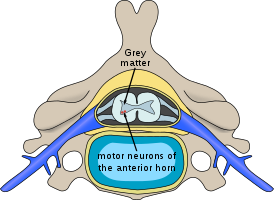
Спина́льная мы́шечная атрофи́я (СМА) (англ. SMA — spinal muscular atrophy) — разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга. Заболевание является аутосомно-рецессивным и связано с мутациями в гене SMN1, кодирующем белок, участвующем в биогенезе сплайсосомы.
Для спинальных мышечных атрофий характерно нарушение работы поперечнополосатой мускулатуры ног, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползание, ходьба, удержание головы, глотание. Мышцы рук обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
Содержание
Общая информация
- СМА — одна из наиболее частых причин детской смертности, вызванной наследственными заболеваниями.
- Спинальные мышечные атрофии детского возраста наследуются по аутосомно-рецессивному типу.
- Ген спинальной мышечной атрофии картирован на 5-й хромосоме, q11.2 — 13.3.
- Ген СМА был идентифицирован в 1995 г., его обозначение SMN (survival motor neuron).
- В среднем один из 6000 — 10000 детей рождается со СМА, в разных странах частота сильно различается.
- 50 % детей с СМА не доживают до двух лет (это дети преимущественно с 1-й формой заболевания).
- SMA может проявиться в любом возрасте, «мягкие» формы проявляются в среднем и пожилом возрасте.
- В среднем каждый 50-й человек имеет рецессивный ген, способный вызывать СМА.
- В соответствии с менделевским расщеплением ребёнок двух носителей поражается СМА с вероятностью 25 %. В этом случае оба родителя несут одиночный дефектный ген, но защищены присутствием второго, нормального гена, который является вообще достаточным для нормальной функции организма. Две дефектных копии гена приводят к генному нарушению, так как не обеспечивается синтез необходимого белка.
- В ходе медико-генетического обследования нескольких российских и среднеазиатских популяций (1,8 млн человек) выявлено 33 больных спинальной мышечной атрофией (СМА): 29 с детской проксимальной СМА (СМА I—III) и 4 c редкими формами. Выявлено «перекрывание» проявлений разных типов СМА I—III (I—II и II—III) у части больных, внутрисемейные различия типов в 3 из 6 семейных случаев, клинико-генетический полиморфизм редких форм СМА. (Г. Е. Руденская, Р. А. Мамедова).
История и патогенез
Спинальная мышечная атрофия у детей впервые была описана Г. Вердниг (G. Werdnig) в 1891 году. Г. Вердниг представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. J. Hoffmann обосновал нозологическую самостоятельность заболевания. В дальнейшем Г. Вердниг и J. Hoffmann (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Е. Kugelberg и L. Welander выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной G. Werdnig и J. Hoffmann.
СМА вызвана мутацией в гене SMN1, который в норме производит белок SMN. Из-за мутации гена, у людей с СМА производится меньшее количество белка SMN, что приводит к потере моторных нейронов.
Классификация типов СМА
Выделяют четыре формы проксимальной спинальной амиотрофии на основе возраста начала, тяжести течения и продолжительности жизни.
| Тип | Эпоним | Обычный возраст начала | Описание | OMIM |
|---|---|---|---|---|
| I: Младенческий | СМА I, болезнь Верднига-Гоффмана | 0-6 мес. | Наиболее неблагоприятная форма СМА. Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием, не держат голову, не сидят самостоятельно. | 253300 |
| II: Промежуточный | СМА II, болезнь Дубовица | 7-18 мес. | Больные этой формой спинальной амиотрофии дети могут есть, сидеть, но никогда не достигают способности ходить самостоятельно. Прогноз в этих случаях зависит от степени вовлечения в патологический процесс респираторных мышц. | 253550 |
| III: Юношеский | СМА III, болезнь Кюгельберга-Веландер | >18 мес. | Наименее опасная форма СМА детского возраста. Пациент способен стоять, но испытывает сильную слабость, с тенденцией к инвалидизации (передвижение в коляске). | 253400 |
| IV: Взрослый | СМА IV или взрослая форма | после 35 лет | Значительно не влияет на продолжительность жизни. Слабость проксимальной мускулатуры, фасцикуляции, снижение сухожильных рефлексов, приводит к неспособности ходить самостоятельно. | 271150 |
Лечение
Радикального лечения не существует.
Так как спинальная мышечная атрофия — нарушение, которое проявляется в синапсах моторных нейронов, состояние может быть улучшено за счёт увеличения уровня SMN — белка. Цель современных исследований — поиск препаратов, увеличивающих уровни SMN. Основные результаты получены пока в исследовательских группах США, Германии, Италии.
Предложено несколько препаратов (вальпроевая кислота, бутират натрия и др.), проводятся их клинические исследования в группах добровольцев. Сведений о результативном применении стволовых клеток пока нет.
Больные СМА нуждаются в специальном диетическом питании, поддерживающей терапии и многих других попечительских действиях.
В декабре 2016 в США было одобрено первое лекарство для лечения СМА — нусинерсен .
Профилактика
Возможна только пассивная профилактика — консультирование родителей с риском СМА о возможных последствиях и пренатальная ДНК-диагностика во время беременности через биопсию ворсин хориона для принятия решения о рождении или прерывании беременности.
Другие формы спинальных мышечных атрофий
Кроме спинальных амиотрофий, обусловленных мутацией в генах SMN1 или SMN2 на длинном плече 5-й хромосомы (5q13.2), вызывающих поражение проксимальных мышц, существует множество схожих заболеваний, большинство из которых — с преимущественным поражением дистальных (то есть ближе к свободному концу конечности) мышц.
| Наименование и синонимы | OMIM | Ген | Локус | Тип наследования | Описание |
| X-сцепленная Спинальная амиотрофия 1-го типа (SMAX1), Spinal and bulbar muscular atrophy (SBMA), Kennedy’s disease (KD) | 313200 | NR3C4 | Xq12 | Х-сцепленный, рецессивный | Позднее начало (в 40-60 лет), медленное прогрессирование, участие в процессе бульбарной группы черепных нервов, нисходящее распространение параличей |
| X-сцепленная Спинальная амиотрофия 2-го типа (SMAX2), Arthrogryposis multiplex congenita — X-linked type 1 (AMCX1) | 301830 | UBA1 | Xp11.23 | Х-сцепленный, рецессивный | Врождённая гипотония и арефлексия вследствие дегенерации и потери двигательных нейронов передних рогов спинного мозга и ствола головного мозга. Часто сочетается с врождёнными контрактурами и/или переломами. Интеллектуальное развитие нормальное. Заболевание быстро прогрессирует, приводя к смерти пациентов до 3-месячного возраста. |
| X-сцепленная Спинальная амиотрофия 3-го типа (SMAX3), Distal spinal muscular atrophy — X-linked (DSMAX) | 300489 | ATP7A | Xq21.1 | Х-сцепленный, рецессивный | Поражены дистальные мышцы всех конечностей, почти всегда у мальчиков, медленно прогрессирующее. |
| Дистальная Спинальная амиотрофия (DSMA1), Spinal muscular atrophy with respiratory distress type 1 (SMARD1), Distal hereditary motor neuropathy type 6 (HMN6) | 604320 | IGHMBP2 | 11q13.3 | Аутосомно-рецессивный | Признаки проявляются с самого рождения, реже во внутриутробном периоде. Заболевание характеризуется преимущественным поражением мышц верхних конечностей и развитием тяжёлых респираторных осложнений из-за прогрессирующей дегенерации мотонейронов передних рогов спинного мозга. |
| Дистальная Спинальная амиотрофия 2-го типа (DSMA2), Distal hereditary motor neuropathy — Jerash type (HMN-J) | 605726 | ? | 9p21.1-p12 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье |
| Дистальная Спинальная амиотрофия 3-го типа (DSMA3), Distal hereditary motor neuropathy types 3 & 4 (HMN3, HMN4) | 607088 | ? | 11q13.3 | Аутосомно-рецессивный | Медленно прогрессирующее |
| Дистальная Спинальная амиотрофия 4-го типа (DSMA4) | 611067 | PLEKHG5 | 1p36.31 | Аутосомно-рецессивный | Медленно прогрессирующее, описано только в одной семье |
| Дистальная Спинальная амиотрофия 5-го типа (DSMA5) | 614881 | DNAJB2 | 2q35 | Аутосомно-рецессивный | Начинается в молодом взрослом возрасте, медленно прогрессирующее. |
| Дистальная Спинальная амиотрофия VA-типа (DSMAVA), Distal hereditary motor neuropathy type 5A (HMN5A) | 600794 | GARS | 7p14.3 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. |
| Дистальная Спинальная амиотрофия VB-типа (DSMAVB), Distal hereditary motor neuropathy type 5B (HMN5B) | 614751 | REEP1 | 2p11 | Аутосомно-доминантный | Преобладает поражение верхних конечностей. |
| Дистальная Спинальная амиотрофия с преимущественным поражением голеней, Distal hereditary motor neuropathy type 2D (HMN2D) | 615575 | FBXO38 | 5q32 | Аутосомно-доминантный | Проявляется в юношестве или у взрослых, медленно прогрессирует, поражает проксимальные и дистальные мышцы, сначала проявляется слабость в голенях, которая распространяется и на руки. |
| Дистальная спинальная амиотрофия с преимущественным поражением голосовых связок, Distal hereditary motor neuropathy type 7A (HMN7A), Harper-Young myopathy. | 158580 | SLC5A7 | 2q12.3 | Аутосомно-доминантный | Проявляется у взрослых параличом голосовых связок, очень редкое заболевание. |
| Аутосомно-доминантная Спинальная амиотрофия, Distal hereditary motor neuropathy type 2A (HMN2A) | 158590 | HSPB8 | 12q24.23 | Аутосомно-доминантный | Проявляется у взрослых. Аллельный вариант болезни Шарко-Мари-Тутса (CMT2L) |
| Аутосомно-доминантная ювенильная Спинальная амиотрофия, Distal hereditary motor neuropathy type 1 (HMN1) | 182960 | ? | 7q34-q36 | Аутосомно-доминантный | Проявляется в юном возрасте |
| Врождённая дистальная Спинальная амиотрофия | 600175 | TRPV4 | 12q24.11 | Аутосомно-доминантный | Поражение двигательных нейронов спинного мозга, иннервирующих нижнюю часть тела. Проявляется непрогрессирующей мышечной атрофией, атрофией мышц бёдер, мышц-разгибателей стопы, слабостью в коленях. Формируются контрактуры в коленных суставах и деформируются стопы. У некоторых пациентов может наблюдаться паралич голосовых связок. |
| Лопаточно-малоберцовая Спинальная амиотрофия (SPSMA), Scapuloperoneal neurogenic amyotrophy | 181405 | TRPV4 | 12q24.11 | Аутосомно-доминантный или Х-сцепленный, доминантный | Поражает мышцы нижних конечностей. Очень редкое заболевание. Аллельный вариант Врождённой дистальная Спинальной амиотрофии. |
| Ювенильная сегментальная Спинальная амиотрофия (JSSMA) | 183020 | ? | 18q21.3 | ? | Начинается в юности, прогрессирует 2-4 года, после чего стабилизируется, влияет в первую очередь на руки, очень редкое. |
| Спинальная амиотрофия Финкеля, Finkel-type proximal spinal muscular atrophy (SMA-FK) | 182980 | VAPB | 20q13.32 | Аутосомно-доминантный | Средний возраст манифестации заболевания 37 лет (известны случаи в возрасте до 12 лет). Симметричная мышечная слабость и истощение мышц. Медленная потеря мышечной силы и прогрессирующая проксимальная атрофия, которая начинается в ногах и со временем распространяется на руки. Также у больных наблюдаются генерализованные фасцикуляции, гипоактивность или отсутствие глубоких сухожильных рефлексов. |
| Спинальная амиотрофия Джокела, Jokela-type spinal muscular atrophy (SMA-J) | 615048 | ? | 22q11.2-q13.2 | Аутосомно-доминантный | Позднее начало, медленно прогрессирование, поражает проксимальные и дистальные мышцы у взрослых. |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 1, Spinal muscular atrophy with lower extremity predominance 1 (SMALED1) | 158600 | DYNC1H1 | 14q32 | Аутосомно-доминантный | Поражает проксимальные мышцы у младенцев. |
| Спинальная амиотрофия с преимущественным поражением нижних конечностей 2, Spinal muscular atrophy with lower extremity predominance 2 (SMALED2) | 615290 | BICD2 | 9q22.31 | Аутосомно-доминантный | Врождённое или с ранним началом, поражающее преимущественно нижние конечности, не прогрессирует, очень редкое. |
| Спинальная амиотрофия с прогрессирующей миоклонической эпилепсией, Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), Jankovic-Rivera syndrome. | 159950 | ASAH1 | 8p22 | Аутосомно-рецессивный | Медленно прогрессирует, преимущественно поражает дистальные мышцы, сочетается с денервацией и миоклоническими приступами. |
| Спинальная амиотрофия с атрофия с врожденными переломами костей, Spinal muscular atrophy with congenital bone fractures (SMA-CBF) | 271225 | ? | ? | Аутосомно-рецессивный? | Тяжёлое истощение мышц (как при болезни Верднига-Гоффмана), сопровождается врожденными переломами костей. |
| Спинальная амиотрофия с понтоцеребеллярной гипоплазией, Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH), Pontocerebellar hypoplasia type 1A (PCH1A) | 607596 | VRK1 | 14q32 | Аутосомно-доминантный | Описано восемь типов понтоцеребеллярной гипоплазии. Частота заболеваний неизвестна. Все формы заболевания имеют общие признаки: аномальное развитие головного мозга, проблемы с двигательной активностью, задержку развития, умственную неполноценность, прогрессирующую микроцефалию и церебральные проявления различной степени. Заболевание проявляется с рождения, в ряде случаев первые признаки отмечаются уже внутриутробно. Пациенты, как правило, погибают в раннем детском возрасте. |
| Ювенильная асимметричная сегментальная Спинальная амиотрофия, Juvenile asymmetric segmental spinal muscular atrophy (JASSMA), Monomelic amyotrophy; Hirayama disease; Sobue disease | 602440 | ? | ? | ? | Болезнь мотонейронов, которая поражает молодых (15-25-летних) мужчин в Индии и Японии. Начинается с мышечной атрофии, которая стабилизируется в плато после 2-5 лет, симптоматика не меняется. Нет боли или потери чувствительности. В отличие от других более низких мотонейронных болезней, MMA, как полагают, не наследуется и редко проявляется фасцикуляциями. |